F2C运营模式
10年+专业及经验
质量可控有保障
服务优质体验
PRODUCT
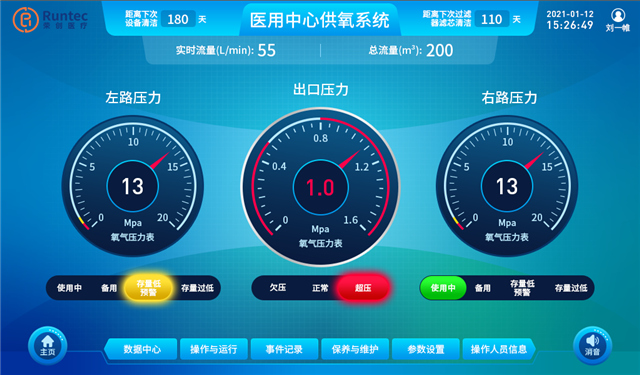
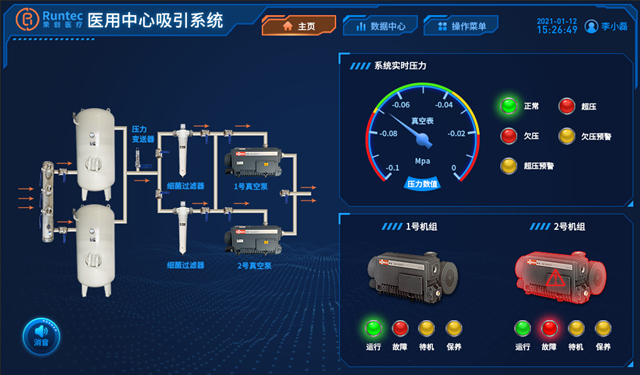
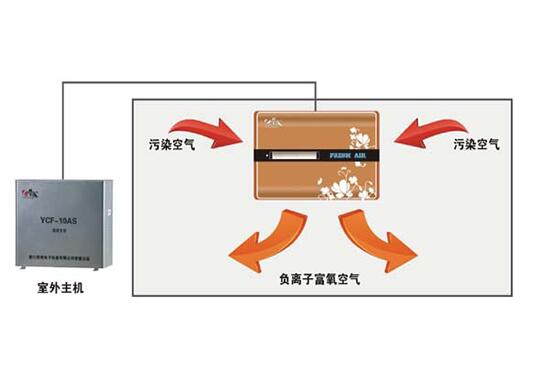
成都中心供氧 产品型号:RC/GY-P-10产品功能特点:采用集中智能控制,实现系统自动运行、故障自动..报警,氧气流量统计(瞬时、月度、累计),超压自动排气;全自动切换氧气汇流排设计,持续供氧;特殊条件下双路同时供氧;可手动操作;大流量供氧、断电持续供氧,恢复供电系统自启动;供氧压力稳定、系统..
服务热线
15756691875
微信